




1989年,中国开始引入医疗器械市场准入的概念,医疗器械新产品需行政审查才可上市;1992年,借鉴欧洲的监管模式,中国启动医疗器械产品安全认证工作;1996年,原国家医药管理局发布《医疗器械产品注册管理办法》,正式规定未经注册的医疗器械不得进入市场;2000年,国务院颁布《医疗器械监督管理条例》(国务院令276号)作为中国的专项行政法规,明确规定实施医疗器械注册管理制度,中国医疗器械监管进入新的发展阶段。
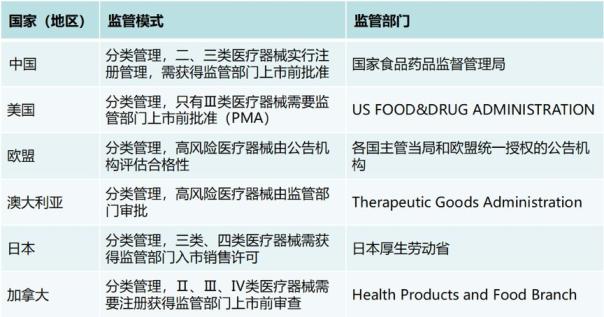
国内外医疗器械监管模式与监管部门对比
2014版《医疗器械监督管理条例》(国务院令650号)于2014年6月1日起施行,体现了国务院关于建立最严格的覆盖全过程的监管制度、深化行政审批制度改革和推进政府职能转变的精神。与旧版条例相比,新条例在完善分类管理、适当减少事前许可、加大企业责任、强化日常监督等方面做出了较大修改,主要特点如下:
1. 明确医疗器械分类管理按照风险高低程度分类;
2. 放开第一类医疗器械的经营,第二类医疗器械经营改为备案;
3. 第一类医疗器械改为备案管理,第二、三类继续实行注册审批管理;
4. “先申请生产许可证后申请产品注册证”改为“先申请产品注册证后申请生产许可证”,减少审批前行政许可事项同时降低企业成本,旧条例规定的16项行政许可降至9项;
5. 强化日常监管职责,增设医疗器械不良事件监测制度、医疗器械延续注册制度、医疗器械召回制度,通过提高处罚幅度、增加处罚种类加大违法行为的处罚力度;
6. 首次明确药监部门和卫生主管部门分别依据各自职责对医疗器械进行监督管理;
7. 明确了申请注册时应提交的资料和文件以及产品注册申请部门,规定第一类医疗器械提交的资料不包括临床试验报告。
除了从上位法加强对医疗器械的监管,我国还发布了创新医疗器械特别审批程序、 医疗器械优先审批程序和免于进行临床试验医疗器械目录等措施提高医疗器械审批效率。也加强了医疗器械上市后和经营、使用环节的监管,2018年监管部门将继续加强医疗器械上市后监管工作,制定一系列规章规范性文件。包括严查网络经营和销售活动,加强医疗器械现场检查并加大处罚力度和改进抽检工作等。围绕保障医疗器械安全有效这一核心目标,监管部门科学提质提速审评审批制度,更细更严监管医疗器械全生命周期。
2018年监管部门计划制定的规章规范性文件

